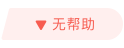编审:Thor
一、导读
共价有机框架(COFs)具有高结晶度、易于化学修饰以及孔径分布可调等优点,是一种有潜力的有机电极材料。此外,其类似聚合物的大分子结构则比有机小分子具备更突出的稳定性,不易溶解在溶剂中。但是,传统的二维COFs仍会通过π-π相互作用堆积成块体,进而导致Li+ 传输距离延长,并且其中的一些活性位点被阻塞在块体 COFs 中。同时,由于合成方法的限制,大多数COFs电极仅包含单一活性的氧化还原基团,这不可避免地导致低的理论和实际容量输出。
因此,实现具有多活性位点的少层 COFs正极材料可能是一种有效的性能提升策略。
二、成果背景
鉴于上述可能有效的研究策略,近日,发表于Advanced Functional Materials上的一篇题为“Dual-Active-Center of Polyimide and Triazine Modified Atomic-Layer Covalent Organic Frameworks for High-Performance Li Storage”的论文实现了该构想。由聚酰亚胺的C=O和三嗪的C=N构筑双活性位点修饰的原子层共价有机框架(E-TP-COF)正极材料改善了锂离子的捕获和扩散:双活性位点有效提升容量、大分子结构难以溶解于电解液。从而实现了110 mAh g-1的高初始容量,并在500次循环后具有高达87.3%的容量保持率。
三、关键创新
四、核心数据解读
1、E-TP-COF的制备
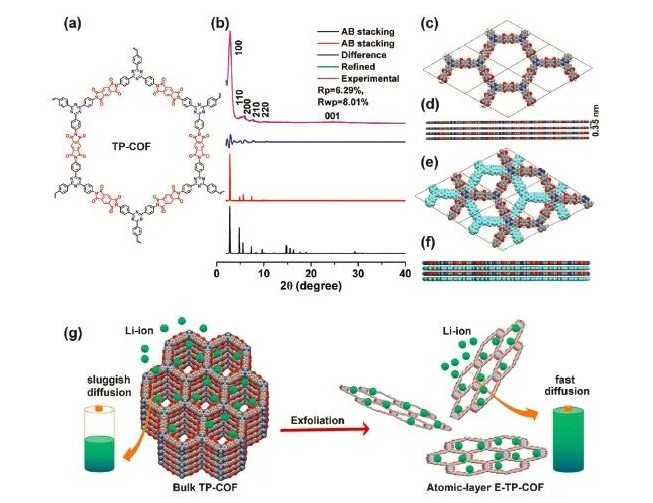
图1:a)双活性中心修饰的TP-COF的化学结构;b)TP-COF的PXRD图谱;c,d)AA堆叠和e,f)AB堆叠的计算模型;g)将TP-COF剥离为E-TP-COF作为锂离子电池正极的示意图。@Wiley
如图1,机械剥离法制备了E-TP-COF。首先,溶液反应得到块状的TP-COF 【1,3,5-三(4-氨基苯基)三嗪(Ta)和均苯四酸二酐(Pm)在N-甲基吡咯烷酮(NMP)、异喹啉和均三甲苯的混合溶液中反应(图1a-f)】。然后,块状TP-COF球磨法机械剥离,得到厚度 ~2.6 nm(相当于14个原子层厚)的少层E-TP-COF(图1g)。
2、E-TP-COF的形貌

图2:a)TP-COF的SEM图像;b)TP-COF的TEM图像;c)TP-COF的HRTEM图像;d)TP-COF和f)E-TP-COF的AFM图像;e)TP-COF和g)E-TP-COF的厚度。@Wiley
如图2a所示,直径~1 µm的球形TP-COF块体剥离后转变为E-TP-COF纳米片。TEM图像中展现了花瓣结构聚集成的球形TP-COF(图2b)。HRTEM标定的层间距为~ 0.35 nm,这与图1d中 的理论值一致,表明TP-COF具有出色的结晶性。AFM的数据显示,剥离前的厚度为~ 60 nm(图2d,e)。 剥离后为~ 2.6 nm(图2f,g)。结合层间距值0.35 nm可得E-TP-COF由大约14个原子层组成。
3、E-TP-COF材料的电化学性能
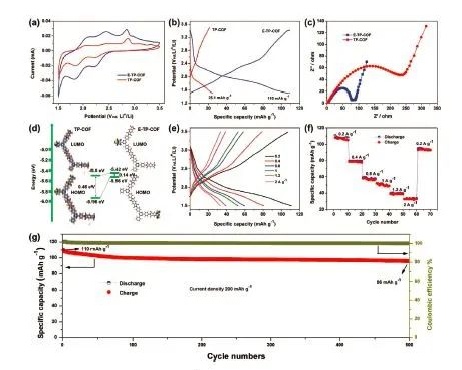
图3:a)E-TP-COF和TP-COF在0.1 mV s-1时的CV曲线;b)E-TP-COF和TP-COF在0.2 A g-1时的恒电流充放电曲线;c)E-TP-COF和TP-COF的阻抗图;d)根据DFT计算得出E-TP-COF和TP-COF的HOMO和LUMO值;e)在不同电流密度下的充放电曲线,以及f)E-TP-COF电极的倍率性能;g)E-TP-COF在0.2 A g-1下的长期循环稳定性。@Wiley
循环伏安数据如图3a所示,TP-COF和E-TP-COF均出现了三对还原/氧化峰。值得一提的是,E-TP-COF的峰值电流远高于TP-COF,源自其暴露和利用了更多的C=O和C=N活性位点。TP-COF和E-TP-COF在电化学性能上的差异,可以通过在200 mA g-1下的恒电流充放电数据来对比。如图3b所示, E-TP-COF电极在2.25和1.85 V处观察到两个放电平台,这归因于C=O和C=N基团的锂化过程, 这与CV结果非常吻合。E-TP-COF则表现出高达110mAh g-1的比容量,远高于TP-COF(25 mAh g-1),这意味着在剥离过程中活性位点的利用率得到明显提高。电化学阻抗谱显示(图3c),相比于块状TP-COF,剥离的E-TP-COF的Rct值更低,表明E-TP-COF电极中的电荷转移动力学优于TP-COF。图3d为TP-COF和E-TP-COF的HOMO和LUMO轨道的能量分布。TP-COF的轨道带隙比E-TP-COF高0.14 eV。E-TO-COF的窄带隙意味着较低的金属离子或电子传输能垒,因此有望获得更好的倍率性能和更高的放电容量。
图3e,f显示了E-TP-COF电极在0.2至2 A g-1电流密度下的倍率性能。可以看出,E-TP-COF电极在0.2 A g-1时可提供超过110 mAh g-1的高初始容量,在2 A g-1时可保持超过30 mAh g-1的容量,远高于TP-COF的对应值。如图3g所示,当电流密度恢复到0.2 A g-1时,E-TP-COF电极在500次循环后,具有87.3%(96 mAh g-1)的容量保持率。
4、E-TP-COF的锂化/脱锂机理研究
图4:a)原位FT-IR光谱设备图和b)E-TP-COF电极在不同的放电和充电状态下的光谱变化。@Wiley
考虑到电池容量高度依赖于活性基团的利用,因此通过FT-IR和XPS对锂化/脱锂过程进行了评估。C=O和C=N基团均可以存储一个Li+并分别形成C-O-Li和C-N-Li。研究表明,在放电/充电过程中,一单位的E-TP-COF可以与总共30个锂离子发生反应。E-TP-COF和锂离子之间的可逆反应过程是通过在不同电位充电/放电过程的FT-IR数据推导出来的(图4a)。在放电过程中(D2.2 V和D1.5 V),三嗪环中的C=O和C=N红外峰强度逐渐降低(1776、1725和1508 cm-1,图4b)。在随后的充电过程中(C2.4和C3.5 V),这些峰的强度明显增加,从而表明电极具有良好的可逆性。
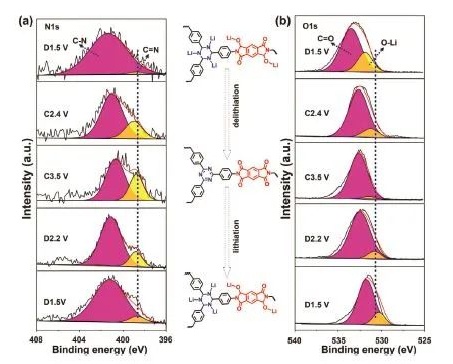
图5:a)E-TP-COF电极在1.5 V下放电、充电至2.4 V和3.5 V以及放电至2.2 V和1.5 V时的N 1s光谱图比较;b)E-TP-COF电极在1.5 V下放电、充电至2.4 V和3.5 V以及放电至2.2 V和1.5 V时的O 1sXPS谱图。 @Wiley
XPS谱图进一步验证了脱锂/锂化过程。如图5a所示,在N1s的光谱中,观察到位于401.4和398.5 eV的两个主峰,分别对应于C-N和C=N。在经过2.4至3.5 V电位下的充电(脱锂过程)后,C=N峰明显增强,表明C-N-Li被还原为C=N基团。但是,再次放电至1.5 V后,C=N峰将再次变宽,表明少层E-TP-COF与锂离子间极好的可逆反应,O1s的XPS光谱也有类似的变化。如图5b所示,放电过程,O1s的XPS谱显示两个主峰,分别对应于E-TP-COF骨架中的C=O键和O-Li键。峰强在充电/放电过程中经历变弱和恢复,表明了可逆的储锂反应。
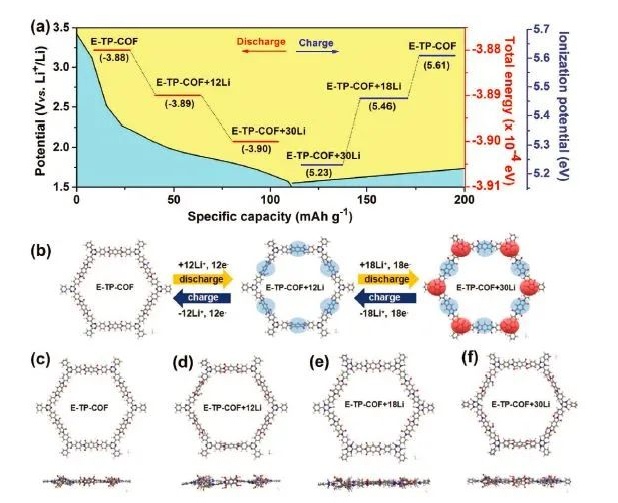
图6 a)E-TP-COF电极的锂化途径。左轴显示了相对于Li+/Li的氧化还原电势,右轴显示不同锂化状态下的E-TP-COF结构的总能量和电离电势。b)通过原子颜色表示在锂化/脱锂过程中的结构演变,白色:H,灰色:C,红色:O,蓝色:N)。Li与C=O和C=N基团之间的结合位点分别由蓝色和红色球体表示。c)E-TP-COF,d)E-TP-COF + 12Li,e)E-TP-COF + 18Li和f)E-TP-COF + 30Li在不同阶段的优化结构示意图。@Wiley
为了深入了解双活性中心修饰的少层E-TP-COF电极材料的多步锂化过程,采用DFT计算模拟了电化学锂存储过程。原则上,E-TP-COF可以在C=O位置接收12个锂离子(E-TP-COF + 12Li),而在C =N位置可以接收18个锂离子(E-TP-COF + 18Li),基于双活性中心概念,E-TP-COF共可以存储30个锂离子(E-TP-COF + 30Li),因此总能量为-3.88×10-4 eV。在放电(锂化)过程中,E-TP-COF + 12Li和E-TP-COF + 30Li的能量分别降低到-3.89×10-4和-3.9×10-4 eV(图6a)。因此,E-TP-COF与E-TP-COF + 12Li之间的总能量之差为 -0.01×10-4 eV。同样的,E-TP-COF和E-TP-COF + 30Li之间的差值为-0.02×10-4 eV,明显小于前者,这意味着Li离子优先与C=O基团的活性位点相互作用。但是,在充电过程中, E-TP-COF + 30Li,E-TP-COF + 18Li和E-TP-COF的电离电势分别为5.23、5.46和5.61 eV(图6a)。较低的电离电势表明更容易失去锂离子。因此,E-TP-COF + 30Li容易丢失12个锂离子以返回E-TP-COF + 18Li状态,紧接着E-TP-COF + 18Li丢失18个锂离子返回初始状态。
五、成果启示
COFs的魅力源自其结构高度可调,因此,构筑多官能团活性位点的策略可以奏效,而剥离又促进了层状结构的形成,确保了电化学性能的释放。
文献链接
欲知成果详情,扫码直达!

声明:本文仅代表作者观点,如有不科学之处,请在下方留言指正!文章系作者授权新威研选发布,转载及相关事宜请联系小威(微信号:xinweiyanxuan)。